Overview
CrassUS is a pipeline for identifying and classifying bacteriophages from the Crassvirales order. Taking as input a metagenome/metavirome, CrassUS is able to identify which contigs are Crassvirales and to classify them into the different families, subfamilies, genera and species of the order.
Flowchart below describes the main steps in the pipeline, which is written in Snakemake for efficiency and reproducibility:
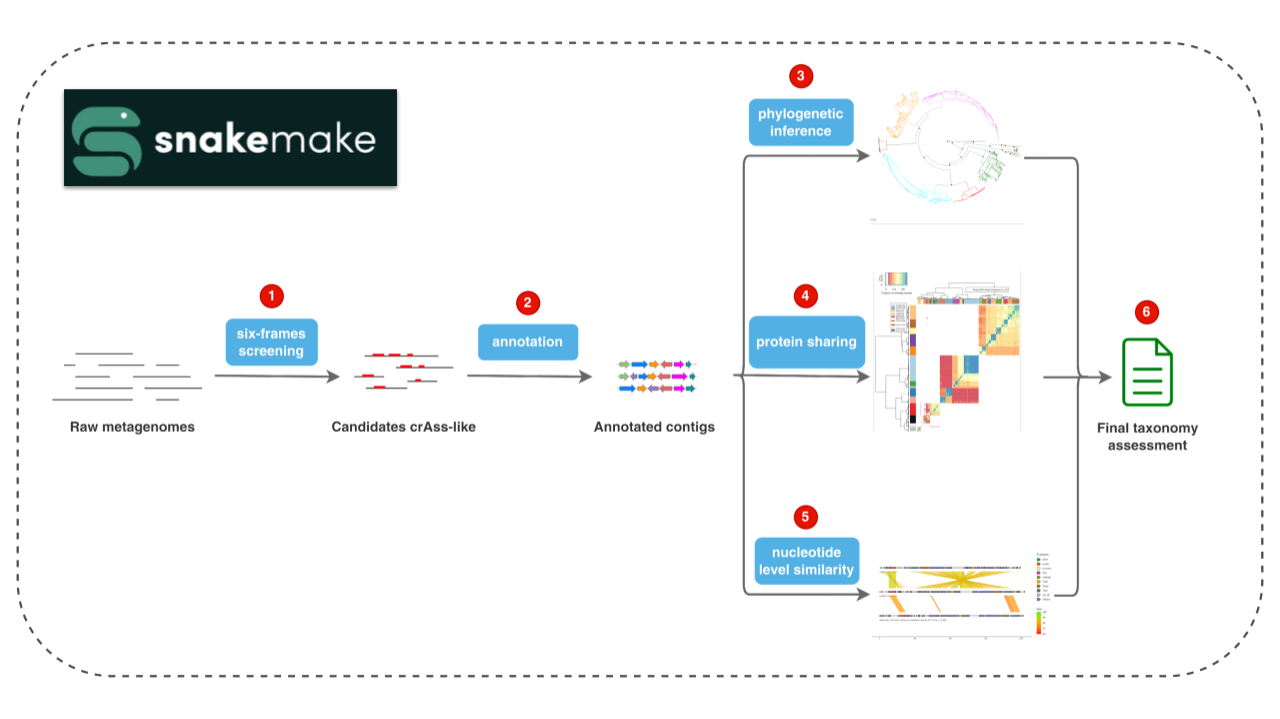
Input contigs are translated into six-frames and scanned with HMM profiles made of conserved, Crassvirales genes sequences such as the large subunit of the terminase (TerL), major capsid protein (MCP) or portal protein. Contigs showing significant similarity to any of these profiles are considered as candidates Crassvirales and analyzed in further steps.
Open Reading Frames (ORFs) are predicted taking into account the opal (TGA) and amber (TAG) stop codon reassignments to tryptophan and glutamine, respectively. ORFs are then functionally characterized by comparing them to a database of conserved Crassvirales profiles.
Phylogenetic relationship with reference Crassvirales is one of the signals used at the classification step. By default, phylogenies for the TerL, MCP and portal proteins are inferred and belonging of candidates to any reference clade is assessed.
The second signal is based on the number of orthologs shared between the candidates and the reference Crassvirales. Cutoffs based on the analysis of solely reference genomes are used to delineate the taxons.
Last signal is based on the similarity at the nucleotide level between the candidates and the reference.
All three signals are combined to annotate the candidates with the deepest possible taxonomic annotation.